Sporadic Creutzfeldt-Jakob Syndrome Misdiagnosed as Recurrent Stroke: A Case Report
Article information

Abstract
While sporadic Creutzfeldt-Jakob disease (sCJD) typically presents with neurological symptoms such as cognitive impairment, ataxia, and myoclonus, its clinical manifestations can be diverse. We report a 70-year-old woman with sCJD who was misdiagnosed with recurrent stroke. She initially showed hemiplegia and high-intensity signals on brain diffusion-weighted magnetic resonance imaging (DWI), which corresponded to the symptoms of a stroke. She was diagnosed with recurrent stroke as her muscle weakness showed stepwise deterioration, with the appearance of additional high-intensity signals on brain DWI. Several days later, she developed Broca’s aphasia, cognitive impairment, and myoclonus in the right upper and left lower extremities. Brain DWI showed high-intensity signals in the cortex, caudate nucleus, and putamen. Therefore, sCJD was suspected; she subsequently underwent further evaluation and was diagnosed with sCJD. The findings of this case indicate that sCJD can have a clinical course similar to that of recurrent stroke.
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) is a fatal neurodegenerative human prion disease. Based on its cause, CJD can be classified as sporadic, iatrogenic, familial, or variable. Most CJD cases (85%) are sporadic, while 10%–15% of cases are familial, and a minority are iatrogenic. The onset of sporadic CJD (sCJD) usually occurs in the seventh decade of life, with a median time to death of 5 months and with 80% of patients dying within 1 year.1,2) sCJD is currently incurable. However, an accurate diagnosis in the early stage is crucial to predict the patient’s prognosis and rule out the diagnosis of other treatable diseases. Here, we report a patient who was misdiagnosed with recurrent stroke based on brain diffusion-weighted magnetic resonance imaging (DWI) findings, which corresponded to hemiplegia, and who had an unusual clinical course that showed stepwise deterioration.
CASE REPORT
A 70-year-old woman with right hemiplegia visited the hospital on the day of onset. She had not experienced any neurological abnormalities before this presentation and had no medical or family history of brain disease. She was a non-smoker, her blood sugar level was normal, and her serum lipid levels were high (total cholesterol, 273 mg/dL; low-density lipoprotein, 184 mg/dL). She was alert, and her cognitive status was good. Her Korean version of the Mini-Mental State Examination score was high (K-MMSE=27), with only three points less than a full score in the attention and calculation category. Her right upper and lower extremity muscle strength measured using the Medical Research Council (MRC) scale was 4/4. No abnormalities were identified on laboratory studies, including those of complete blood count, serum electrolytes, liver function, and renal function on the day of admission. Brain DWI showed hyperintense signals in the left frontal and temporal lobe areas (Fig. 1A). Based on hemiplegic symptoms and corresponding brain imaging results, the patient was diagnosed with cerebral infarction and treatment with aspirin, clopidogrel, and atorvastatin was initiated. Until the 12th day of hospitalization, the patient’s symptoms did not worsen or disappear, although her muscle strength remained weakened.
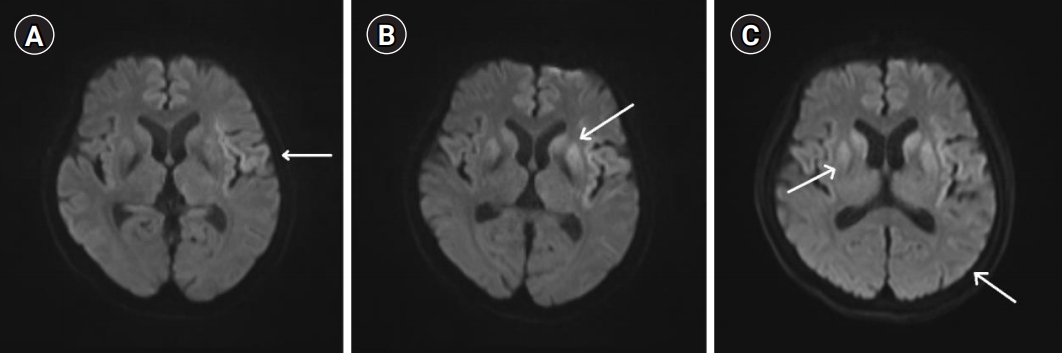
Axial brain diffusion-weighted images. (A) Initial axial brain diffusion-weighted magnetic resonance imaging (DWI) showing high-intensity signals (arrow) in the left frontal and temporal lobes (0 days from symptom onset). (B) Axial brain DWI showing additional high-intensity signals (arrow) in the left frontal and temporal lobes and the caudate nucleus and putamen (12 days from symptom onset). (C) Axial brain DWI showing extension of the high-intensity signals (arrows) to both the cortex and caudate nucleus and the putamen (18 days from symptom onset).
On the 12th day of hospitalization, the patient’s level of consciousness temporarily changed to drowsy and her right upper and lower extremity muscle strength measured using the MRC scale deteriorated by 1/2. Brain DWI revealed additional hyperintense signals in the left frontal and temporal lobes and both basal ganglia areas, which led to a diagnosis of recurrent cerebral infarction and continued treatment for stroke (Fig. 1B).
On the 18th day of hospitalization, the patient developed Broca’s aphasia. Her cognition deteriorated, and myoclonus appeared in her right upper and left lower extremities. Her scores for memory recall, calculation, and orientation in K-MMSE dropped, with her overall score also lower than the previous results scores (K-MMSE score=20). Repeated brain DWI confirmed that the hyperintense signals extended to both the cortex and caudate nucleus and putamen (Fig. 1C). In addition to rapidly progressive cognitive impairment, myoclonus, and pyramidal symptoms, she had hyperintense signals in both the caudate nucleus and putamen on brain DWI. Therefore, she was strongly suspected of having CJD and was transferred to another university hospital for accurate diagnostic assessment.
Western blot analysis detected 14-3-3 proteins in her cerebrospinal fluid; real-time quaking-induced conversion assays also detected the scrapie isoform of the prion protein in the cerebrospinal fluid. The possibility of familial CJD was ruled out as no genetic mutations associated with CJD were found in the prion genetic test and she had no family history of CJD. The possibility of iatrogenic CJD was also excluded because she had no medical history of neurosurgical treatment, surgery, or other body tissue transplant at risk of transmission of prion proteins. Thus, the patient was diagnosed with sCJD. The informed consent was waived.
DISCUSSION
The most common early symptom of sCJD is cognitive impairment, and other symptoms such as cerebellar, constitutional, behavioral, sensory, motor, and visual symptoms may also appear.3,4) The symptoms of sCJD can vary and appear in unusual patterns; thus, this condition may be easily mistaken for other central nervous system disorders.5) Therefore, the best approach in suspected cases of sCJD is to consider patient care with a critical appraisal of the information known about the disease. The diagnostic criteria for sCJD are outlined in Table 1.6)
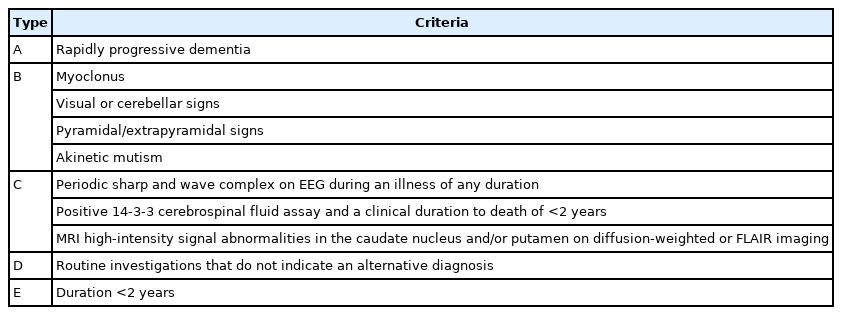
Diagnostic criteria for sCJD
While the clinical features of sCJD can be diverse, acute onset and stepwise deterioration of the pyramidal symptoms resembling the clinical course of a recurrent stroke are rare. However, cerebrovascular diseases usually worsen in a stepwise pattern; thus, it is rare to see a gradual deterioration of symptoms over several hours or days.7) A previous study reported a case in Korea in which hemiplegia occurred as an initial symptom, with corresponding brain magnetic resonance imaging (MRI) results, after which the patient was diagnosed with sCJD. The case exhibited progressive deterioration of symptoms; the typical symptoms of sCJD such as rigidity, bradykinesia, and tremor appeared 8 days later, which enabled the early suspicion of sCJD.7) Another case was reported in which atypical white matter lesions were observed on brain MRI, but the initial symptoms were typical of sCJD.8) Another case in Germany described a patient with hemiplegia and corresponding brain MRI findings. This case showed symptoms of cognitive decline before hemiplegia onset, followed by rapid cognitive deterioration. The patient further showed deterioration of symptoms such as dystonic-hyperkinetic movement and myoclonus, which are symptoms of sCJD.9) Such cases attained a relatively early diagnosis of sCJD as the symptoms had a gradual progressive onset and did not resemble the clinical course of a recurrent stroke.
The patient in the present report had cerebrovascular risk factors such as hypercholesterolemia, old age, and menopause. She also presented with hemiplegia and corresponding high-intensity signals on brain MRI; moreover, her symptoms had acute onset and deteriorated in a stepwise pattern. Furthermore, the changes identified on brain MRI corresponded to those for stroke, leading to a diagnosis of recurrent stroke. Consequently, antiplatelet therapy continued until the typical symptoms of sCJD appeared. The clinical course and imaging results of this case, which resembled those of recurrent stroke, demonstrated the variability of sCJD symptoms. Therefore, it is crucial to understand the potential for uncommon clinical outcomes of sCJD to avoid unnecessary testing and treatment.
Notes
CONFLICT OF INTEREST
The researchers claim no conflicts of interest.
FUNDING
None.
AUTHOR CONTRIBUTIONS
Conceptualization, MJB; Data curation, MJB; Methodology, MJB, YRH; Project administration, MJB, YRH; Supervision, KHH; Writing–original draft, MJB, IHK; Writing–review & editing, MJB, KHH.