INTRODUCTION
In mammals, the Sestrin (Sesn) family of stress-sensitive genes comprises three members: Sesn1, Sesn2, and Sesn3.1) Sesns have dual biochemical functions: first, they act as antioxidants that control the activity of peroxiredoxins (PRXs),2) in which the expression level of Sesn2 is critical for redox homeostasis. The ectopic expression of Sesn2 significantly reduces reactive oxygen species (ROS) levels and increases cell viability, whereas the suppression of Sesn2 increases intracellular ROS levels and reduces cell viability following hydrogen peroxide exposure.2) Sesns also function as inhibitors of target of rapamycin complex 1 (TORC1) signaling.3) Drosophila Sesn (dSesn) has been reported to prevent age-associated pathologies, including fat accumulation and cardiac and skeletal muscle degeneration, by promoting a feedback loop that prevents excessive TORC1 activation and ROS accumulation.3) As ROS accumulation4) and TORC1 activation5,6) are associated with accelerated aging and the development of age-associated pathologies in both invertebrates and vertebrates, we tested whether the loss of Sesn2 induces cellular senescence in mouse embryonic fibroblasts (MEFs) before investigating Sesn2 knockout (KO) mice at the organismal level.
Cellular senescence is an irreversible growth arrest state provoked by diverse stresses.4) In particular, the relationship between cellular senescence and ROS was confirmed by the observation that treatment with exogenous hydrogen peroxide triggers certain primary cells to quickly enter senescence.7-9) The results of the present study showed that Sesn2 KO induced cellular senescence in MEFs and demonstrated that Sesn2 was an important regulator of cellular senescence via NADPH oxidase 4 (NOX4)-dependent ROS generation and subsequent activation of AMP-activated protein kinase (AMPK).
MATERIALS AND METHODS
Mice
Sesn2-KO mice were produced by transferring two-cell embryos deficient in Sesn2 into the oviduct of a foster mother. The Sesn2-KO embryos were provided by the Mutant Mouse Resource & Research Centers at the University of California, Davis. Wild-type (WT) and Sesn2-KO mice with the 129/SvJ background were maintained in a pathogen-free authorized facility at the Korea Research Institute of Bioscience and Biotechnology (KRIBB) under a 12-hour dark/light cycle at 20°C–22°C and 50%–60% humidity. All animal procedures were conducted according to the guidelines of the Institutional Animal Care and Use Committee of KRIBB.
Preparation of MEFs
MEFs were prepared from 13.5-day-old embryos derived from Sesn2-KO mice mated with each other. The head, tail, and viscera were removed, and the remaining body was minced, dispersed in 0.25% trypsin/EDTA, and incubated in 5% CO2 at 37°C for 30 minutes. Large fragments were removed, and the cell suspensions were plated on 10-cm plates and incubated at 37°C until confluent.
Cell Culture
WT and Sesn2 KO MEFs, as well as 293T cells, were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 20 mM HEPES, and antibiotics (Life Technologies Corp., Carlsbad, CA, USA) at 37°C in a humidified atmosphere containing 5% CO2.
Immunoblotting
Immunoblotting was performed as described previously.9) Briefly, cells were lysed in lysis buffer (20 mM HEPES, pH 7.2, 50 mM NaCl, 0.5% Triton X-100, 10% glycerol, 1 µg/mL aprotinin, 1 µg leupeptin, 1 mM Na3VO4, and 1 mM NaF). Antibodies against the following proteins were used: sirtuin 1 (SIRT1), p16Ink4a, p21Cip1, α-actinin, human influenza hemagglutinin (HA), and β-actin (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA); phospho-Smad3, phospho-AMPK, phospho-Akt, p53, and AMPK (all from Cell Signaling Technology Inc., Danvers, MA, USA); Sesn2 (ProteinTech Group Inc., Chicago, IL, USA); nucleoredoxin (R&D Systems Inc., Minneapolis, MN, USA); α-tubulin (Calbiochem, San Diego, CA, USA); γ-tubulin (Abcam Inc., Cambridge, MA, USA); paired related homeobox 1 (PRX1), PRX2, and PRX3; anti-thioredoxin (TRX; a kind gift from Dr. Ho Zoon Chae, Chonnam National University, Gwangju, Korea); and anti-malate dehydrogenase 1 (MDH1; a kind gift from Dr. Hong-Duk Youn, Seoul National University, Seoul, Korea).
Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated using RNA-spin (Intron Biotechnology Inc., Seongnam, Korea). cDNA was synthesized from 1 μg of total RNA using a DiaStar RT Kit (SolGent, Gwanpyeong-dong, Korea). The primers are listed in Table 1. The housekeeping genes β-actin and GAPDH were used as endogenous controls for normalization.
Determination of ROS Production
The 2ʹ,7ʹ-dichlorofluorescein diacetate (DCFH-DA; Life Technologies Corp.) oxidation-sensitive probe was used to measure intracellular ROS levels by flow cytometry. Briefly, cells were incubated with 10 μM DCFH-DA for 30 minutes. For flow cytometry analysis, the cells were detached by trypsinization, washed once in phosphate-buffered saline (PBS), and resuspended in 800 μL PBS. Flow cytometric analyses (10,000 events per sample) were performed using a FACSCalibur system (BD Biosciences, San Jose, CA, USA) with excitation and emission wavelengths of 485 nm and 538 nm, respectively, and evaluated using CellQuest software.
SA-β-Gal Activity
Senescence-associated β-galactosidase (SA-β-Gal) staining was performed as previously described.10) Briefly, the cells were washed in PBS, fixed in 2% formaldehyde and 0.2% glutaraldehyde for 5 minutes, and washed three times. Next, the cells were incubated in fresh SA-β-Gal stain solution—1 mg/mL X-gal (5-bromo-4-chloro-3-indolyl b-D-galactoside) stock (20 mg/mL in dimethylformamide)/40 mM citric acid/sodium phosphate, pH 6.0/5 mM potassium ferrocyanide/5 mM potassium ferricyanide/150 mM NaCl2—for 12–16 hours at 37°C. Blue cells were counted under an inverted microscope (Axiovert25; Carl Zeiss, Oberkochen, Germany).
Cell Proliferation Assay
Relative cell proliferation was measured as previously described.9) Briefly, WST-1 solution (DoGen, Seoul, Korea) was added to the cells for 2 hours, and the absorbance was measured at 450 nm using a VICTOR3 Multilabel Plate Reader (PerkinElmer Inc., Waltham, MA, USA). The cells were then treated with the TGF-β type I receptor kinase inhibitor SB431542 (SB; 5 µM, Calbiochem), the NOX inhibitor diphenyleneiodonium (DPI; 5 µM, Calbiochem), and the AMPK inhibitor compound C (5 µM, Sigma-Aldrich, St. Louis, MO, USA) for 48 hours after plating.
Transfection
The 293T cells were transfected with the HA-mCherry human Sesn2 (hSesn2) and SBE4 reporter plasmid, pretreated with TGF-β (2 ng/mL) for 24 hours, and subjected to a reporter assay. Relative luciferase activity was analyzed using a Promega Luciferase Assay System (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions.
RESULTS
Loss of Sesn2 Induces Cellular Senescence
To determine the role of Sesn2 in cellular senescence, we examined the effect of Sesn2-KO on senescence in MEFs. MEFs isolated from Sesn2-KO mice exhibited obvious cellular senescence phenotypes (Fig. 1A). We observed SA-β-Gal-positive cells among Sesn2-KO MEFs but not among WT MEFs (Fig. 1A, 1B). The Sesn2-KO MEFs also became flattened and enlarged (Fig. 1A), constituting the morphological changes characterizing senescent cells. Next, we examined whether Sesn2-KO prevented cell proliferation. Compared with WT MEFs, Sesn2-KO MEFs showed a 25% decrease in proliferation (Fig. 1C). These results suggested that Sesn2 was involved in the progression of cellular senescence. However, despite increased cellular senescence, there were no obvious differences in the basal levels of p16Ink4a and p21Cip1, which induce cell cycle arrest and accelerated senescence,11) in Sesn2-KO MEFs compared to WT MEFs (Fig. 1D).
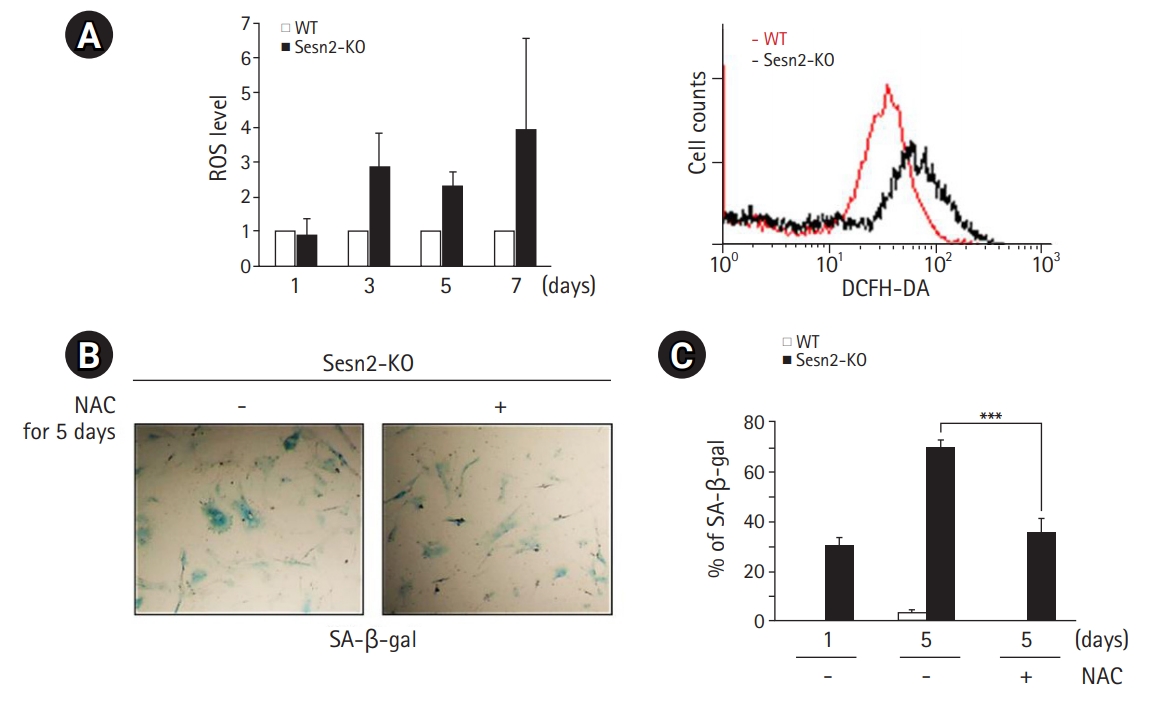
Loss of Sesn2 Generates ROS
The characteristics of Sesn deficiency have been reported previously in flies.3) To investigate the mechanisms by which the loss of Sesn2 induces cellular senescence in MEFs, we measured intracellular ROS levels in Sesn2-KO MEFs by staining with DCFH-DA, a peroxide-sensitive fluorescent probe. DCFH-DA passively enters the cell, where it reacts with ROS to form the highly fluorescent compound dichlorofluorescein. As shown in Fig. 2A, the fluorescence intensity of dichlorofluorescein was significantly increased in Sesn2-KO MEFs compared to that in WT MEFs on days 3, 5, and 7 after plating. To explore the effect of ROS on cellular senescence, we analyzed whether changes in ROS levels affected cellular senescence in Sesn2-KO MEFs. Treatment with the antioxidant N-acetyl-cysteine (NAC) significantly decreased the SA-β-Gal activity in Sesn2-KO MEFs (Fig. 2B, 2C). These results suggested that the loss of Sesn2 accelerated cellular senescence via ROS generation.
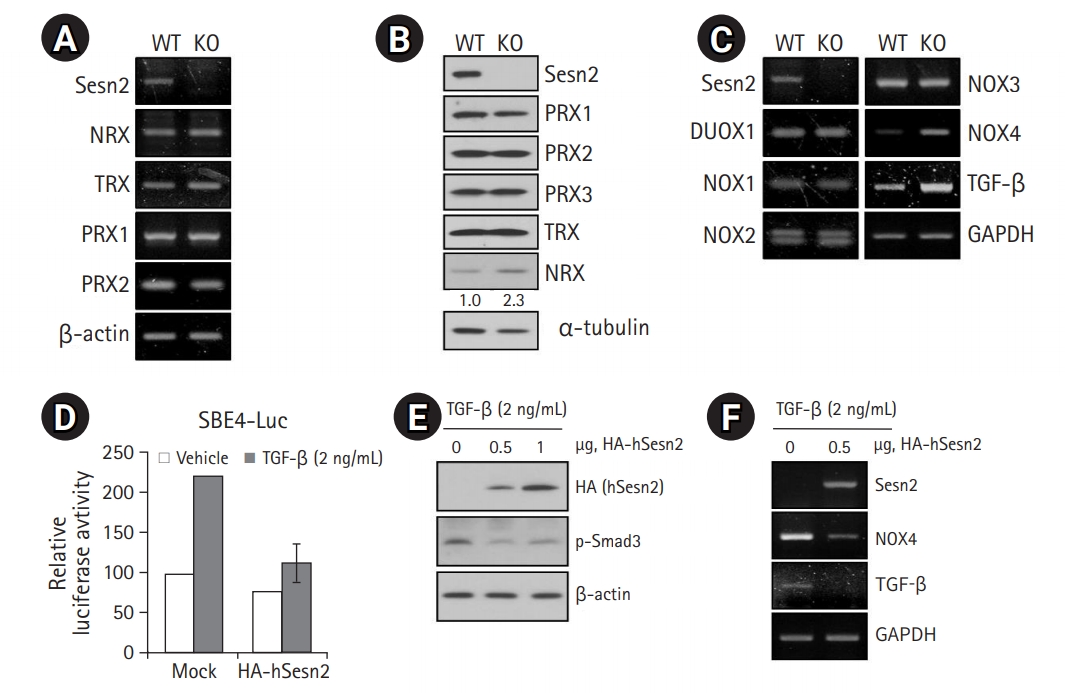
Loss of Sesn2 Triggers NOX4
An increase in ROS levels by lowering antioxidant levels reportedly accelerates cellular senescence, whereas an increase in ROS scavenging delays senescence.12) Therefore, we measured the levels of antioxidant proteins in Sesn2-KO MEFs to determine whether the loss of Sesn2 increases ROS levels by regulating the expression of antioxidant enzymes. No obvious changes were observed in the mRNA expression levels and protein abundance of several antioxidants, including PRX1, PRX2, PRX3, and TRX, between WT and Sesn2-KO MEFs (Fig. 3A, 3B), suggesting that cellular senescence induced by loss of Sesn2 was not mediated by a decrease in antioxidant levels. However, NRX protein levels were slightly increased in Sesn2-KO MEFs (Fig. 3B).
As NOX family members generate ROS,13) we investigated which NOX family members were involved in Sesn2 signaling. We analyzed the expression patterns of DUOX1, NOX1, NOX2, NOX3, and NOX4 in WT and Sesn2-KO MEFs by RT-PCR. We observed markedly increased NOX4 expression in Sesn2-KO MEFs (Fig. 3C), suggesting that NOX4 played an important role in ROS production in Sesn2-KO MEFs.
Because TGF-β upregulates NOX4,14,15) we measured the mRNA levels of TGF-β to determine whether the loss of Sesn2 increased TGF-β levels. We observed increased mRNA levels of TGF-β in Sesn2-KO MEFs (Fig. 3C). Next, we induced the overexpression of hSesn2 in 293T cells. We found that hSesn2 expression markedly decreased TGF-β promoter activity (Fig. 3D), Smad3 phosphorylation (Fig. 3E), and mRNA levels of TGF-β, ultimately leading to a decrease in NOX4 mRNA levels (Fig. 3F). Together, these results indicated that Sesn2 is a key regulator of the TGF-β-mediated NOX4 pathway.
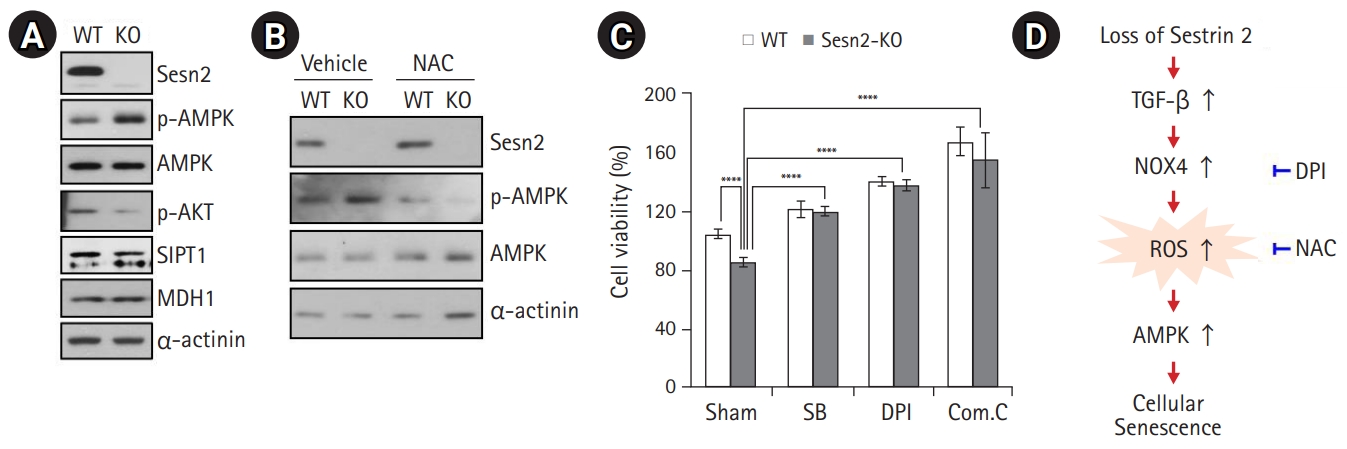
AMPK Activation is Involved in the Loss of Sesn2-Induced Senescence
Abundant evidence indicates that a rise in intracellular ROS levels contributes to cellular senescence.4,12) Moreover, ROS also induces ATP depletion.16) AMPK is an energy sensor that is activated by increased levels of intracellular AMP. Generally, AMPK activation turns on catabolic pathways that generate ATP while also inhibiting cell proliferation and biosynthetic processes that consume ATP. Thus, we hypothesized that elevated ROS levels lead to AMPK activation in Sesn2-KO MEFs, likely via a decline in ATP levels. We found that the loss of Sesn2 led to AMPK activation in MEFs with a reduction in Akt phosphorylation and SIRT1 protein levels (Fig. 4A). As MDH1 knockdown induces senescence in human fibroblasts,17) we examined whether MDH1 was related to senescence in Sesn2-KO MEFs. No changes were observed in the protein levels of MDH1 in Sesn2-KO MEFs (Fig. 4A). To clarify the effect of ROS on AMPK activation, we treated Sesn2-KO MEFs with NAC and observed significantly suppressed AMPK activation (Fig. 4B). We then investigated the proliferation of Sesn2-KO MEFs using the TGF-β type I receptor kinase inhibitor SB, the NOX inhibitor DPI, and the AMPK inhibitor compound C. SB, DPI, and compound C treatment significantly increased the growth of Sesn2-KO MEFs compared to that of WT MEFs (Fig. 4C), indicating that the TGF-β/NOX4/AMPK pathway may contribute to proliferation defects in Sesn2-KO MEFs (Fig. 4D).
DISCUSSION
The results of this study allowed us to characterize Sesn2 as a crucial regulator of cellular senescence. Sesn2 was first reported as a disulfide reductase of PRX,2) and dSesn has been shown to prevent excessive ROS accumulation.3) In contrast, one study reported that Sesn2 does not function as a reductase of cysteine sulfinic acid in PRXs.18)
We observed that Sesn2-KO MEFs generated ROS. Loss of Sesn2 has been associated with the induction of TGF-β signaling independent of ROS accumulation.19) TGF-β has also been shown to upregulate NOX4, which produces ROS.14,15) NOX4 overexpression also induces cellular senescence.20) On the basis of these reports, we evaluated whether TGF-β and NOX4 were involved in ROS generation in association with the senescence phenotype in Sesn2-KO MEFs. RT-PCR analysis revealed that NOX4 is the major NADPH oxidase isoform expressed in Sesn2-KO MEFs (Fig. 3C), suggesting that the loss of Sesn2 induces TGF-β-mediated NOX4 expression.
Cellular senescence participates in four complex biological processes (tumor suppression, tumor promotion, aging, and tissue repair), some of which have opposing effects.21) Furthermore, cellular senescence is triggered by a complex signaling network involving the interaction of multiple proteins, including those associated with mitochondrial function, ROS, senescence, and chromatin remodeling.17,22)
Lowered ATP production and subsequently an elevated AMP:ATP ratio and increased AMPK activity are associated with age.23) Moreover, ROS can activate AMPK via decreased ATP levels.24) Evidence suggests that AMPK plays a role in cellular senescence. Activated AMPK leads to phosphorylation of p53 at serine 15, which induces p53-dependent cellular senescence.25) Activated AMPK has also been shown to inhibit the RNA-binding protein HuR.23) HuR levels are reduced in senescent cells and HuR overexpression restores a young phenotype, whereas a reduction in HuR expression accentuates senescence-associated morphology.26) Finally, activated AMPK contributes to the inhibition of mammalian target of rapamycin (mTOR) signaling by activation of the tuberous sclerosis complex 2 (TSC2) in response to energy stress.27) Activation of TSC2 by AMPK strongly suppresses cell proliferation.28) In addition, AMPK directly phosphorylates mTOR at threonine 2446 following stimulation with insulin, thereby inhibiting mTOR action.29) Thus, AMPK can inhibit mTOR activity to limit cell proliferation as well as protein synthesis both directly and indirectly (via TSC2).
Both mRNA and protein levels of SIRT1 are markedly decreased during senescence; a decrease in SIRT1 levels leads to the acetylation of liver kinase B1 (LKB1), which, in turn, induces AMPK-dependent senescence. Moreover, the phosphorylation of Akt at serine 473 is markedly decreased in senescent cells. The inhibition of Akt induces cellular senescence, and liver kinase B1 overexpression markedly decreases Akt activity when endogenous SIRT1 is inhibited.30) Although we did not elucidate the detailed signaling cascade downstream of AMPK, we observed slightly decreased SIRT1 protein levels and significantly inhibited phosphorylation of Akt at serine 473 in Sesn2-KO MEFs compared to those in WT MEFs (Fig. 4A). Our results showed that Sesn2 was a key factor of senescence via AMPK activation. This role of Sesn2 is likely mediated by a NOX4-mediated increase in ROS levels, as NAC prevented the increase in ROS levels and attenuated AMPK activation (Fig. 4B). We also found that inhibitor-mediated abrogation of TGF-β and NOX4 activities resulted in increased cell proliferation (Fig. 4C). Our results suggest a novel Sesn2-mediated pathway that may underlie the induction of NOX4, generation of ROS, and subsequent activation of AMPK in cellular senescence.